
Bạn có biết rằng nhiều người thường nhầm lẫn giữa Alpha và Beta Thalassemia? Cả hai đều là bệnh tan máu di truyền nhưng mỗi thể bệnh lại có cơ chế di truyền, mức độ nghiêm trọng và tiên lượng hoàn toàn khác nhau. Bài viết dưới đây sẽ giúp bạn so sánh bệnh Alpha Thalassemia và Beta Thalassemia, từ đó hiểu rõ đặc điểm của từng thể bệnh để chủ động trong tầm soát, chẩn đoán, điều trị và phòng ngừa nhóm bệnh này hiệu quả..
Tổng quan về bệnh Thalassemia
Thalassemia là nhóm bệnh tan máu di truyền lặn trên nhiễm sắc thể thường, đặc trưng bởi sự giảm hoặc mất khả năng tổng hợp các chuỗi globin (thành phần quan trọng cấu tạo nên hemoglobin trong hồng cầu). Bệnh xảy ra khi gen chịu trách nhiệm tổng hợp chuỗi alpha globin hoặc beta globin bị đột biến, làm mất cân bằng số lượng các chuỗi trên, từ đó gây rối loạn sản xuất hemoglobin và làm tế bào hồng cầu dễ bị phá vỡ hơn, dẫn đến tình trạng thiếu máu mạn tính.
Về cơ chế bệnh sinh, trong hemoglobin bình thường, mỗi phân tử Hb cấu tạo từ 2 chuỗi alpha và 2 chuỗi beta globin:
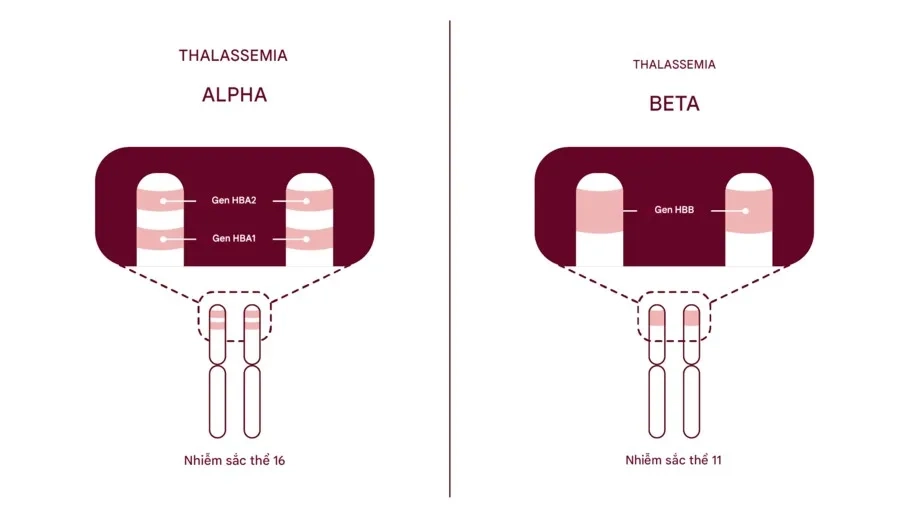
- Chuỗi Alpha globin: Được tổng hợp từ 2 gen HBA1 và 2 gen HBA2 nằm trên cặp nhiễm sắc thể số 16.
- Chuỗi Beta globin: Được tổng hợp từ 2 gen HBB nằm trên cặp nhiễm sắc thể số 11.
Khi thiếu hụt một trong hai loại chuỗi globin, số lượng loại chuỗi globin còn lại bị dư thừa và tự tạo thành các phân tử hemoglobin bất thường kết tụ trong tế bào hồng cầu, gây tổn thương và phá huỷ hồng cầu sớm. Điều này dẫn tới hai hậu quả: khiến cơ thể thiếu máu kéo dài và buộc tủy xương phải hoạt động quá mức để bù lại và lắng đọng các chất trung gian từ hồng cầu vỡ ở các mô và cơ quan, từ đó dẫn đến các biến chứng nghiêm trọng như biến dạng xương, gan lách to và tổn thương chức năng gan thận.
Sự thiếu hụt chuỗi alpha globin gây ra bệnh Alpha Thalassemia, trong khi thiếu hụt chuỗi beta globin dẫn đến bệnh Beta Thalassemia. Tuỳ thuộc vào mức độ rối loạn tổng hợp các chuỗi này, người bệnh sẽ có biểu hiện từ nhẹ đến nặng.
Đặc điểm của Alpha Thalassemia
Bệnh Alpha Thalassemia xảy ra do đột biến gây ảnh hưởng đến chức năng của gen HBA1 và/hoặc HBA2, hai gen chịu trách nhiệm sản xuất chuỗi alpha globin của hemoglobin, gồm bốn bản sao nằm trên cặp nhiễm sắc thể số 16.
Tuỳ vào số lượng bản sao gen bị mất hoặc đột biến, Alpha Thalassemia được phân thành bốn thể bệnh chính, gồm:
- Alpha Thalassemia thể ẩn (mất 1 gen): Đây là tình trạng người lành mang gen bệnh và không biểu hiện triệu chứng lâm sàng. Người bệnh vẫn khỏe mạnh, sinh hoạt bình thường và chỉ được phát hiện qua xét nghiệm sàng lọc và xét nghiệm gen.
- Alpha Thalassemia thể nhẹ (mất 2 gen): Thường gây thiếu máu nhẹ và không cần điều trị.
- Alpha Thalassemia thể HbH (mất 3 gen): Biểu hiện lâm sàng vô cùng đa dạng, có thể gây thiếu máu mức độ trung bình đến nặng, thường đi kèm với các triệu chứng như gan lách to, vàng da, chậm phát triển thể chất và có thể cần truyền máu định kỳ.
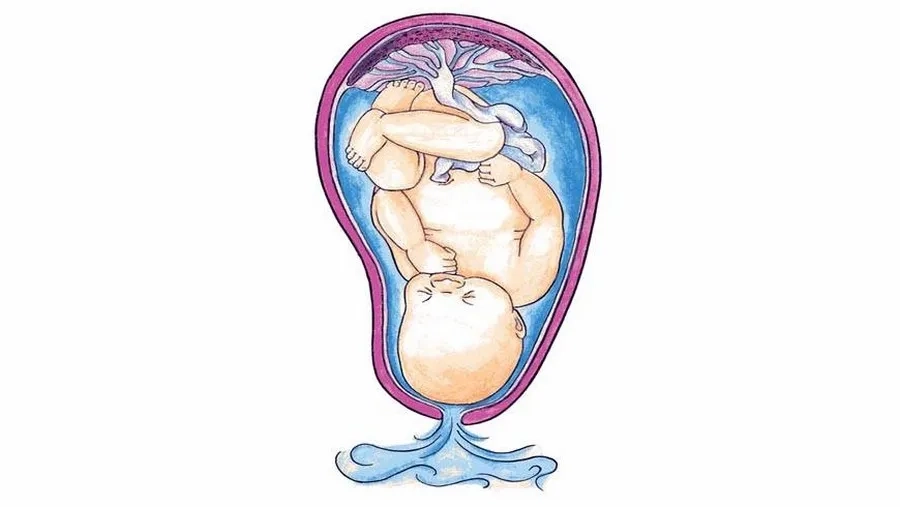
- Alpha Thalassemia thể phù thai Bart’s (mất 4 gen): Đây là thể nặng nhất, xảy ra do đột biến mất hoàn toàn bốn gen alpha-globin, dẫn đến không hình thành hemoglobin chức năng. Điều này gây nên tình trạng phù thai nghiêm trọng và đa số các trường hợp tử vong trong tử cung hoặc ngay sau sinh.
Mặc dù đã được phân loại rõ ràng theo các hướng dẫn quốc tế, việc tiên lượng bệnh Alpha Thalassemia vẫn còn nhiều khó khăn do có nhiều loại đột biến khác nhau và tương tác các gen, các bệnh lý liên quan. Để có cái nhìn khách quan, đa chiều, cá thể hóa, việc tư vấn với chuyên gia di truyền ở mỗi người bệnh, người mang gen là vô cùng cần thiết.
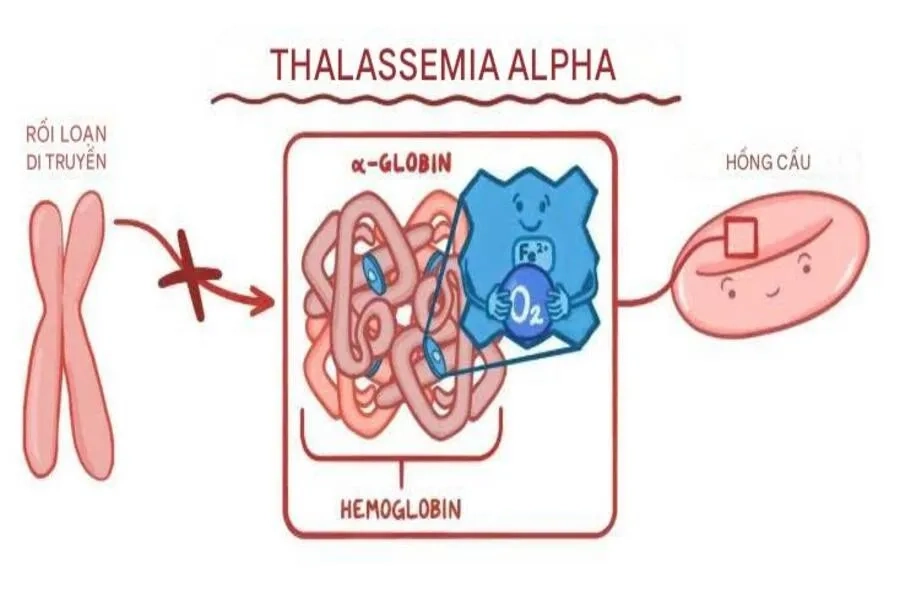
Sơ đồ cơ chế bất thường di truyền của bệnh Alpha Thalassemia
Đặc điểm của Beta Thalassemia
Bệnh Beta Thalassemia xảy ra do đột biến gây ảnh hưởng đến chức năng của gen HBB, gen chịu trách nhiệm sản xuất chuỗi beta globin của hemoglobin, gồm hai bản sao nằm trên cặp nhiễm sắc thể số 11.
Dựa vào mức độ suy giảm tổng hợp chuỗi beta, bệnh được chia thành 3 thể chính:
- Thể mang gen: Người bệnh chỉ mang một gen đột biến, thường không có triệu chứng hoặc chỉ thiếu máu nhẹ. Thể bệnh này thường không cần điều trị, người mang gen sống khỏe mạnh bình thường, nhưng vẫn có thể truyền gen bệnh cho con nếu kết hôn cùng người cũng mang gen đột biến
- Thể trung gian: Xảy ra khi người bệnh có hai gen HBB bị tổn thương mức độ vừa phải (thường là do một gen mất chức năng hoàn toàn và một gen giảm chức năng, hoặc cả hai đều giảm chức năng). Thể bệnh này thường gây tình trạng thiếu máu nhẹ đến vừa và có thể cần truyền máu rải rác. Người mắc thể bệnh này nếu không được điều trị cũng có thể gặp các triệu chứng như: gan lách to, biến dạng xương, loãng xương, sỏi mật và rối loạn nội tiết.
- Thể nặng: Đây là thể bệnh nghiêm trọng nhất, xảy ra khi cả hai gen HBB đều bị đột biến mất chức năng hoàn toàn. Bệnh thường biểu hiện sớm từ trong năm đầu với biểu hiện thiếu máu nặng và cần truyền máu định kỳ suốt đời để duy trì sự sống. Nếu không được điều trị đúng cách, người bệnh dễ gặp các biến chứng như biến dạng xương mặt, chậm phát triển thể chất, suy giảm chức năng nhiều cơ quan nội tạng và nguy cơ tử vong sớm.
Biểu hiện của bệnh Beta Thalassemia rất đa dạng, phụ thuộc vào kiểu đột biến gen, mức độ suy giảm tổng hợp chuỗi beta globin và sự tương tác với các biến thể di truyền khác. Đặc biệt, với nhiều đột biến điểm đa dạng, người bệnh Beta Thalassemia có khả năng xuất hiện nhiều loại hemoglobin bất thường trong máu (huyết sắc tố bất thường), gây khó khăn lớn trong quá trình tiên lượng bệnh.
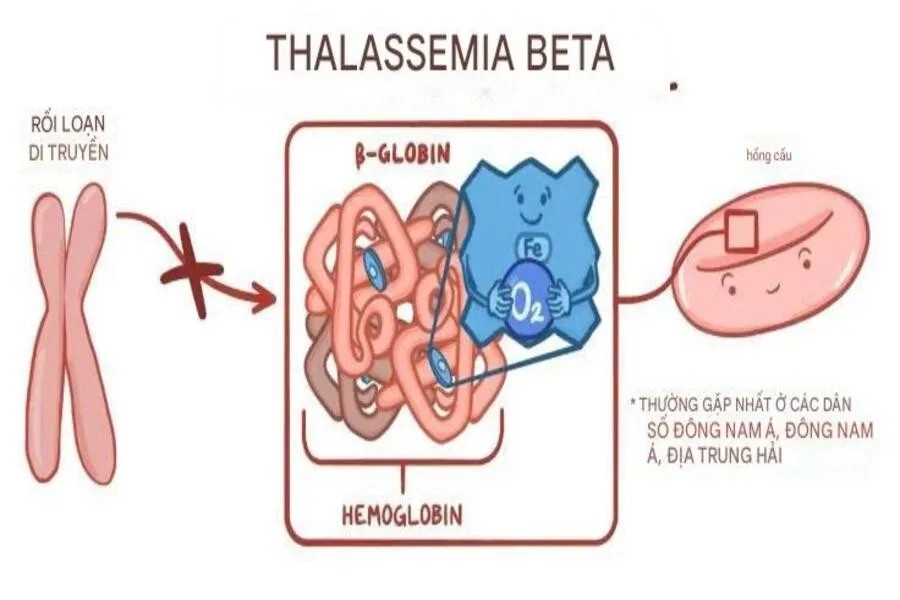
Sơ đồ cơ chế bất thường di truyền của bệnh Beta Thalassemia
So sánh bệnh Alpha Thalassemia và Beta Thalassemia
Việc phân tích hai thể Alpha và Beta Thalassemia giúp làm rõ sự tương đồng trong cơ chế bệnh sinh cũng như những khác biệt đáng kể về nguyên nhân di truyền, thời điểm biểu hiện bệnh và tiên lượng lâm sàng. Hiểu đúng bản chất của từng thể không chỉ hỗ trợ bác sĩ trong quá trình chẩn đoán và điều trị, mà còn đóng vai trò thiết yếu trong công tác tư vấn di truyền và dự phòng trước sinh, nhất là tại các vùng có tỷ lệ mang gen cao.
1. Điểm giống nhau
Cả hai thể Thalassemia Alpha và Beta đều là bệnh lý có cơ chế di truyền lặn trên nhiễm sắc thể thường, liên quan đến sự rối loạn tổng hợp chuỗi globin (thành phần thiết yếu của hemoglobin trong hồng cầu).
Về cơ chế bệnh sinh, cả hai đều là hệ quả của sự thiếu hụt chuỗi alpha hoặc beta globin, dẫn đến mất cân bằng trong tổng hợp hemoglobin, làm tổn thương hồng cầu và gây tình trạng thiếu máu mạn tính.
Biểu hiện lâm sàng của hai thể bệnh này có nhiều điểm tương đồng, đều gây thiếu máu mạn tính với mức độ từ nhẹ đến nặng, gây dư thừa sắt tích lũy tại các cơ quan và biểu hiện các triệu chứng như da xanh xao, mệt mỏi, gan lách to, vàng da, chậm phát triển thể chất và biến dạng xương nếu không được phát hiện sớm và điều trị kịp thời.
Đặc biệt, cả hai đều không lây nhiễm, nhưng có khả năng di truyền sang con nếu cả bố và mẹ đều mang gen bệnh.
2. Sự khác biệt
Mặc dù cùng là rối loạn di truyền ảnh hưởng đến quá trình tổng hợp hemoglobin, Alpha Thalassemia và Beta Thalassemia khác nhau rõ rệt về nguyên nhân di truyền, thời điểm biểu hiện bệnh và tiên lượng. Dưới đây là một số cách phân biệt Alpha Thalassemia và Beta Thalassemia dựa trên 3 đặc điểm sau:
2.1. Gen tổn thương
Bệnh Alpha Thalassemia xảy ra do đột biến gen HBA1 và HBA2 trên nhiễm sắc thể số 16.
Bệnh Beta Thalassemia xảy ra do đột biến gen HBB nằm trên nhiễm sắc thể số 11.
2.2. Thời điểm biểu hiện
Bệnh Alpha Thalassemia có thể gây hậu quả nghiêm trọng ngay trong thời kỳ bào thai. Thể nặng nhất (phù thai Bart’s) có thể làm thai nhi có biểu hiện thiếu máu, phù thai và tử vong ngay từ trong bụng mẹ do không tổng hợp được chuỗi alpha globin - một thành phần cần thiết của phân tử HbF (hemoglobin bào thai). Trong khi đó, ở Beta Thalassemia, thể nặng nhất thường bắt đầu biểu hiện khi trẻ được từ 6 tháng tuổi trở lên, thời điểm HbF giảm dần và HbA không được tổng hợp đầy đủ.
2.3. Mức độ nghiêm trọng và tiên lượng
Ở bệnh Alpha Thalassemia, thể nặng nhất có thể gây tử vong sớm trong thai kỳ và ảnh hưởng đến cả mẹ trong quá trình mang thai (gây phù thai, tiền sản giật, sản giật...). Trong khi đó, bệnh Beta Thalassemia thể nặng cho phép trẻ sống sót sau sinh, nhưng lại cần truyền máu và điều trị thải sắt suốt đời hoặc ghép tủy để duy trì sự sống. Biến chứng thường gặp bao gồm: gan lách to, loãng xương, rối loạn nội tiết, suy tim hoặc xơ gan do ứ sắt.
Alpha Thalassemia và Beta Thalassemia cái nào nặng hơn?
Trên thực tế, không thể đưa ra câu trả lời duy nhất về cái nào nặng hơn khi so sánh Thalassemia thể Alpha và Beta, bởi mức độ trầm trọng của từng thể bệnh còn phụ thuộc vào kiểu gen tổn thương, mức độ thiếu hụt chuỗi globin, thời điểm phát hiện cũng như khả năng tiếp cận điều trị.
Tuy nhiên, nếu so sánh ở cấp độ bào thai, bệnh Alpha Thalassemia thể phù thai (mất cả 4 gen alpha) được xem là nghiêm trọng nhất, vì có thể gây phù thai nặng, suy tim thai và dẫn đến chết lưu. Đồng thời, nó cũng làm tăng nguy cơ biến chứng sản khoa nguy hiểm cho mẹ như tiền sản giật, đa ối và băng huyết.
Ngược lại, nếu nhìn ở góc độ diễn tiến sau sinh và chất lượng sống lâu dài, bệnh Beta Thalassemia thể nặng là một bệnh lý mạn tính nghiêm trọng, khiến người bệnh phải truyền máu định kỳ và điều trị thải sắt suốt đời hoặc phải ghép tủy điều trị. Đồng thời, người mắc bệnh cũng phải đối mặt với nhiều biến chứng nặng nề và nguy cơ tử vong sớm nếu không được quản lý tốt.
Người mắc Thalassemia thể nặng phải truyền máu suốt đời để duy trì sự sống
Kết luận
So sánh bệnh Alpha Thalassemia và Beta Thalassemia không chỉ giúp phân biệt rõ đặc điểm di truyền, mức độ nghiêm trọng và hướng điều trị của từng thể bệnh, mà còn là bước quan trọng trong chiến lược sàng lọc, phòng ngừa hiệu quả các bệnh lý huyết học bẩm sinh. Dù Alpha hay Beta Thalassemia, khi không được phát hiện sớm và kiểm soát đúng cách, đều có thể dẫn đến những hậu quả nghiêm trọng, ảnh hưởng đến sức khỏe và chất lượng sống của người bệnh.
Để được tư vấn di truyền, xét nghiệm tầm soát và theo dõi điều trị bệnh Thalassemia một cách bài bản, bạn có thể đến Bệnh viện Đại học Phenikaa (PhenikaaMec). Với đội ngũ chuyên gia huyết học, sản, nhi và di truyền học giàu kinh nghiệm, cùng hệ thống xét nghiệm công nghệ cao, bệnh viện cung cấp dịch vụ tư vấn di truyền, sàng lọc trước hôn nhân, trước sinh, chăm sóc thai kỳ nguy cơ cao và điều trị Thalassemia theo hướng cá thể hóa. Hãy liên hệ hotline 1900 886648 để đặt lịch thăm khám cùng bác sĩ chuyên khoa.